Yuyang Lei, Jiao Zhang, Cara R. Schiavon https://orcid.org/0000-0002-9311-2145, Ming He, Lili Chen, Hui Shen, Yichi Zhang, Qian Yin, Yoshitake Cho, Leonardo Andrade, Gerald S. Shadel, Marcos Hepokoski, Ting Lei, Hongliang Wang, Jin Zhang, Jason X.-J. Yuan, Atul Malhotra, Mansão Uri https://orcid.org/0000-0002-9802-1955, Shengpeng Wang, Zu-Yi Yuan [email protected], e John YJ. Tímido
A infecção por SARS-CoV-2 (síndrome respiratória aguda grave coronavírus 2) depende da ligação da proteína S (glicoproteína Spike) à ECA (enzima conversora de angiotensina) 2 nas células hospedeiras. O endotélio vascular pode ser infectado pelo SARS-CoV-2, o que desencadeia a produção de espécies reativas de oxigênio mitocondriais e a mudança glicolítica. Paradoxalmente, a ECA2 é protetora no sistema cardiovascular, e a proteína S do SARS-CoV-1 promove lesão pulmonar ao diminuir o nível de ECA2 nos pulmões infectados. No estudo atual, mostramos que a proteína S sozinha pode danificar células endoteliais vasculares (ECs) ao regular negativamente a ECA2 e, consequentemente, inibir a função mitocondrial.
Administramos um pseudovírus expressando proteína S (Pseu-Spike) a hamsters sírios intratraquealmente. Danos pulmonares foram aparentes em animais que receberam Pseu-Spike, revelados pelo espessamento dos septos alveolares e aumento da infiltração de células mononucleares (Figura [A]). AMPK (proteína quinase ativada por AMP) fosforila ACE2 Ser-680, MDM2 (duplo minuto murino 2) ubiquitina ACE2 Lys-788, e a interação entre AMPK e MDM2 determina o nível de ACE2. Nos pulmões danificados, os níveis de pAMPK (fosfo-AMPK), pACE2 (fosfo-ACE2) e ACE2 diminuíram, mas os de MDM2 aumentaram (Figura [B], i). Além disso, a fosforilação complementar aumentada e diminuída de eNOS (NO sintase endotelial) Thr-494 e Ser-1176 indicou atividade eNOS prejudicada. Essas alterações na expressão de pACE2, ACE2, MDM2 e atividade de AMPK no endotélio foram recapituladas por experimentos in vitro usando células-tronco arteriais pulmonares infectadas com Pseu-Spike que foi resgatado pelo tratamento com N-acetil-L-cisteína, um inibidor de espécies reativas de oxigênio (Figura [B], ii).
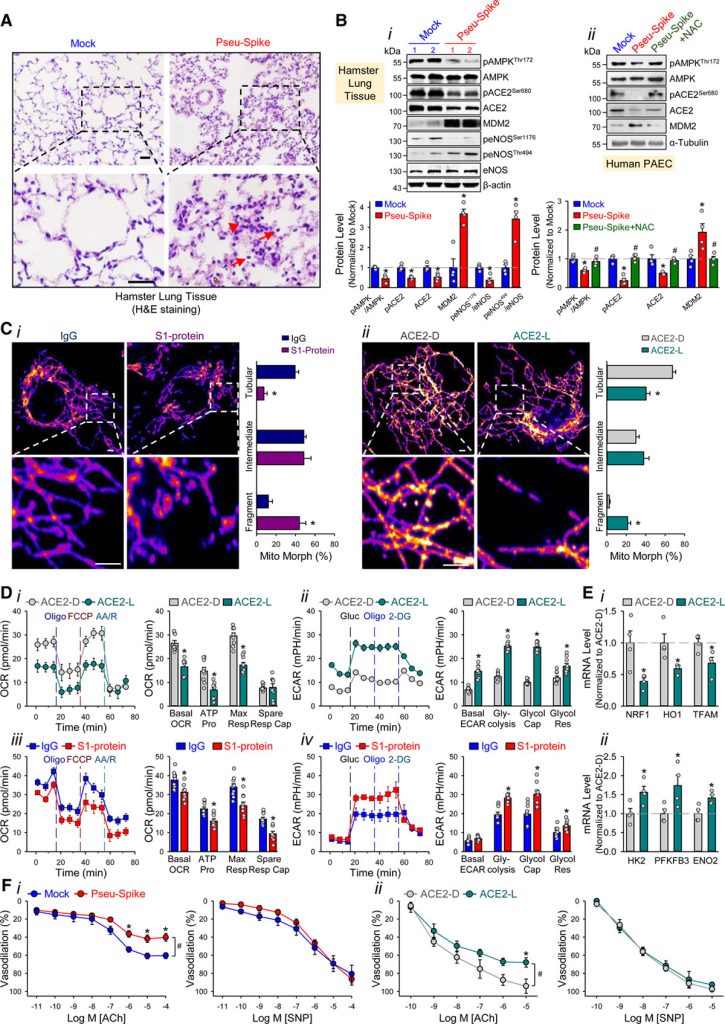
Em seguida, estudamos o impacto da proteína S na função mitocondrial. Imagens confocais de ECs tratadas com proteína S1 revelaram aumento da fragmentação mitocondrial, indicando dinâmica mitocondrial alterada (Figura [C], i). Para examinar se essas alterações mitocondriais eram devidas, em parte, à quantidade reduzida de ACE2, superexpressamos ACE2 S680D (ACE2-D, um ACE2 fosfo-mimético com estabilidade aumentada) ou S680L (ACE2-L, um defosfo-mimético com estabilidade reduzida) em ECs. Conforme mostrado na Figura [C], ii, ECs com ACE2-L tiveram um número maior de mitocôndrias fragmentadas quando comparadas àquelas com ACE2-D. Ao realizar ensaios de taxa de consumo de oxigênio e taxa de acidificação extracelular, descobrimos que ECs superexpressando ACE2-L tiveram respiração mitocondrial basal, produção de ATP e respiração máxima reduzidas em comparação com ECs superexpressando ACE2-D (Figura [D], i). Além disso, a superexpressão de ACE2-L causou aumento na taxa de acidificação basal, glicólise induzida por glicose, capacidade glicolítica máxima e reserva glicolítica (Figura [D], ii). Além disso, ECs incubadas com proteína S1 tiveram função mitocondrial atenuada, mas aumentaram a glicólise, quando comparadas com células de controle tratadas com IgG (Figura [D], iii e iv). Também comparamos as expressões de genes relacionados à mitocôndria e à glicólise em ECs pulmonares isoladas de camundongos knock-in de ACE2-D ou ACE2-L. Mostrado na Figura [E], os níveis de mRNA de NRF1 , HO1 e TFAM (genes relacionados à biogênese da mitocôndria) foram aumentados, enquanto aqueles de HK2 , PFKFB3 e ENO2 (genes relacionados à glicólise) foram diminuídos em ECs pulmonares em camundongos ACE2-D, em comparação com aqueles em camundongos ACE2-L.
A infecção por SARS-CoV-2 induz inflamação da CE, levando à endotelite. Como a proteína S diminuiu o nível de ACE2 e prejudicou a biodisponibilidade de NO, examinamos se a entrada da proteína S é indispensável para o endotélio disfuncional. Conforme mostrado na Figura [F], i, a vasodilatação dependente do endotélio induzida pela acetilcolina foi prejudicada em artérias pulmonares isoladas de hamsters administrados com Pseu-Spike, enquanto a vasodilatação independente do endotélio induzida pelo nitroprussiato de sódio não foi afetada. Também comparamos a vasodilatação induzida por acetilcolina e nitroprussiato de sódio de vasos pulmonares de camundongos ACE2-D ou ACE2-L. Conforme previsto, a vasodilatação induzida pela acetilcolina foi prejudicada em artérias pulmonares isoladas de camundongos ACE2-L em comparação com camundongos ACE2-D (Figura [F], ii). No entanto, houve pouca diferença na vasodilatação induzida por nitroprussiato de sódio entre os animais ACE2-D e ACE-L.
Embora o uso de um pseudovírus não infeccioso seja uma limitação deste estudo, nossos dados revelam que a proteína S sozinha pode danificar o endotélio, manifestado por função mitocondrial prejudicada e atividade eNOS, mas aumento da glicólise. Parece que a proteína S em ECs aumenta o estresse redox, o que pode levar à desativação de AMPK, regulação positiva de MDM2 e, finalmente, desestabilização de ACE2. Embora essas descobertas precisem ser confirmadas com o vírus SARS-CoV-2 no estudo futuro, parece paradoxal que a redução de ACE2 pela proteína S diminuiria a infectividade do vírus, protegendo assim o endotélio. No entanto, um sistema renina-angiotensina desregulado devido à redução de ACE2 pode exacerbar a disfunção endotelial, levando à endotelite. Coletivamente, nossos resultados sugerem que o dano de EC exercido pela proteína S anula a infectividade diminuída do vírus. Esta conclusão sugere que o anticorpo gerado pela vacinação e/ou anticorpo exógeno contra a proteína S não apenas protege o hospedeiro da infectividade do SARS-CoV-2, mas também inibe a lesão endotelial imposta pela proteína S.
lei-et-al-2021-sars-cov-2-spike-protein-impairs-endothelial-function-via-downregulation-of-ace-2
Fonte: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.121.318902