Abstrato
O reposicionamento de drogas, a abordagem de descobrir diferentes usos para drogas existentes, ganhou enorme popularidade nos últimos anos no campo de descoberta de drogas anticancerígenas devido à crescente demanda por drogas anticancerígenas. Além disso, o reaproveitamento de medicamentos antiparasitários veterinários para o tratamento do câncer está ganhando força, conforme apoiado pela literatura existente. Um exemplo proeminente é a proposta de implementar o uso de antiparasitários veterinários, como os carbamatos de benzimidazole e as salicilanilidas halogenadas, como novos medicamentos anticancerígenos. Esses agentes têm revelado atividades antitumorais pronunciadas e ganharam atenção especial pelo “duplo reposicionamento”, pois são reaproveitados para diferentes espécies e doenças simultaneamente, atuando por diferentes mecanismos dependendo de seu alvo. Como agentes anticancerígenos, esses compostos empregam vários mecanismos, incluindo a inibição das vias de transdução de sinal oncogênico da respiração mitocondrial e a inibição das respostas celulares ao estresse. Nesta revisão, resumimos e fornecemos informações valiosas sobre os ensaios experimentais, pré-clínicos e clínicos de medicamentos antiparasitários veterinários disponíveis para o tratamento de vários tipos de câncer em humanos. Esta revisão sugere a possibilidade de novas opções de tratamento que possam melhorar a qualidade de vida e os resultados para pacientes com câncer em comparação com os tratamentos atualmente utilizados
Palavras-chave: reaproveitamento de medicamentos, antiparasitários, carbamatos de benzimidazol, salicilanilidas halogenadas, terapia do câncer
1. Introdução
O câncer é uma doença extensa e a causa mais comum de morbidade e mortalidade em todo o mundo. É caracterizada por uma desregulação do ciclo celular, que resulta principalmente em uma perda progressiva do controle do crescimento e diferenciação celular. Embora existam numerosos estudos em andamento sobre terapia anticancerígena, com muitos candidatos principais em várias fases de pesquisa pré-clínica ou clínica, apenas 5% das possíveis terapias anticancerígenas que entram em ensaios clínicos de fase I foram aprovadas e entraram no mercado. Os tratamentos padrão para o câncer incluem cirurgia, imunoterapia, radiação e quimioterapia. Atualmente, a quimioterapia é uma das estratégias mais eficientes e potentes utilizadas no tratamento de tumores malignos. No entanto, o desenvolvimento de multirresistência aos quimioterápicos tornou-se um grande impedimento para o sucesso do tratamento do câncer. Claramente, novas alternativas terapêuticas são necessárias para melhorar o diagnóstico e o tratamento do câncer. Antes de serem comercializados como um novo medicamento, os compostos principais enfrentam muitos obstáculos durante os estudos pré-clínicos e clínicos para garantir sua qualidade, segurança, dosagem e eficácia. Os ensaios clínicos são caros e demorados, exigindo de dez a quinze anos de pesquisa dedicada. Todo o processo de desenvolvimento de colocar um único composto candidato no mercado é dificultado pelos custos exorbitantes (aproximadamente US$ 1 a US$ 2 bilhões) associados aos testes necessários para a aprovação da Food and Drug Administration (FDA) dos EUA.
O reaproveitamento de medicamentos ganhou reconhecimento na última década, permitindo que os produtos farmacêuticos existentes fossem reconsiderados para aplicações alternativas. Reduziu o risco de um medicamento não chegar ao mercado, devido à baixa carga de efeitos adversos, à atenuação da carga econômica e à agilidade do processo de aprovação. Também pode oferecer uma melhor relação entre risco e recompensa, pois reduz o cronograma do processo de desenvolvimento de medicamentos e também é economicamente viável quando comparado a outras estratégias de desenvolvimento de medicamentos. Além disso, os resultados pré-clínicos obtidos com o uso de medicamentos reaproveitados podem acelerar o processo de tradução pré-clínica para clínica do tratamento do câncer.
Exemplos de medicamentos reconhecidos como agentes de alto potencial dentro do projeto Repurposing Drugs in Oncology incluem cimetidina, claritromicina, diclofenaco, mebendazol (MZ) e nitroglicerina, entre outros. Um número considerável de drogas investigadas para reaproveitamento em oncologia são antiparasitários e já estão em uso clínico contra diferentes vermes parasitas há várias décadas. Os medicamentos antiparasitários são um grupo de candidatos que atuam contra os vermes parasitas que colonizam o intestino. Essas drogas foram originalmente desenvolvidas para tratar parasitas veterinários e, posteriormente, ganharam espaço para aplicações clínicas em pacientes humanos. Potenciais candidatos farmacológicos incluem carbamatos de benzimidazol (BZ) e salicilanilidas halogenadas (HS), que têm sido amplamente utilizados como medicamentos antiparasitários veterinários e, posteriormente, foram reaproveitados para o tratamento do câncer humano. Isso lançou as bases para o fenômeno do “duplo reposicionamento”, que pode ser definido como o reposicionamento de um medicamento existente para tratar diferentes doenças e espécies ao mesmo tempo. As atividades anticancerígenas dos anti-helmínticos foram relatadas para MZ, pamoato de pirvinio e niclosamida (Nic) por Mukhopadhyay et al. em 2002, Esumi et al. em 2004, e Wang et al. em 2009, respectivamente. Desde então, vários estudos relataram os efeitos desses compostos contra células tumorais in vitro e in vivo, e em ensaios clínicos de fase 1. Esta revisão resume as evidências e informações atuais sobre a atividade anticancerígena das drogas do grupo BZ e HS em linhagens celulares, modelos de tumores animais e ensaios clínicos, que podem ajudar a melhorar a qualidade de vida de pacientes com câncer.
2. Processo de Seleção de Artigos
Utilizamos os mecanismos de busca PubMed e Google Scholar para reunir uma lista de publicações e manuscritos que investigam a atividade anticancerígena do carbamato BZ e HS em linhagens celulares, modelos de tumores animais e ensaios clínicos. Para que um relatório seja incluído nesta pesquisa, ele deve conter carbamato BZ e HS no título ou no resumo. As palavras-chave “BZ carbamato” e “HS”, combinadas com “albendazol”, “mebendazol”, “fenbendazol”, “flubendazol”, “ricobendazol”, “niclosamida”, “closantel”, “rafoxanida” ou “atividade anticancerígena” foram usados para gerar a lista. Qualquer artigo de revisão foi excluído de nossa pesquisa. Artigos altamente relevantes foram inicialmente determinados pelo título e resumo, seguido por um exame mais aprofundado para confirmar se a pesquisa conduzida com os grupos de drogas BZ carbamato e HS foi em linhas celulares ou em seres humanos ou animais. Reconhecemos que esta técnica de busca não foi enciclopédica, pois existem muitos artigos de periódicos que não constam no PubMed ou Google Scholar. Avaliamos os estudos escolhidos avaliando diferentes características, como o tipo de espécie, a fonte celular, as linhagens celulares, o tipo de câncer e a via alvo.
3. Carbamatos BZ
Os antiparasitários BZ são um grupo de compostos orgânicos aromáticos heterocíclicos amplamente utilizados em medicamentos humanos e veterinários para inibir parasitas internos. Alguns medicamentos BZ importantes incluem MZ, albendazol (ABZ), fenbendazol (FZ), flubendazol (FLU), triclabendazol, parbendazol, oxibendazol e ricobendazol. Nos últimos anos, alguns deles foram investigados com sucesso para vários tipos de câncer em todo o mundo.
3.1. Mecanismo de Ação dos Carbamatos BZ
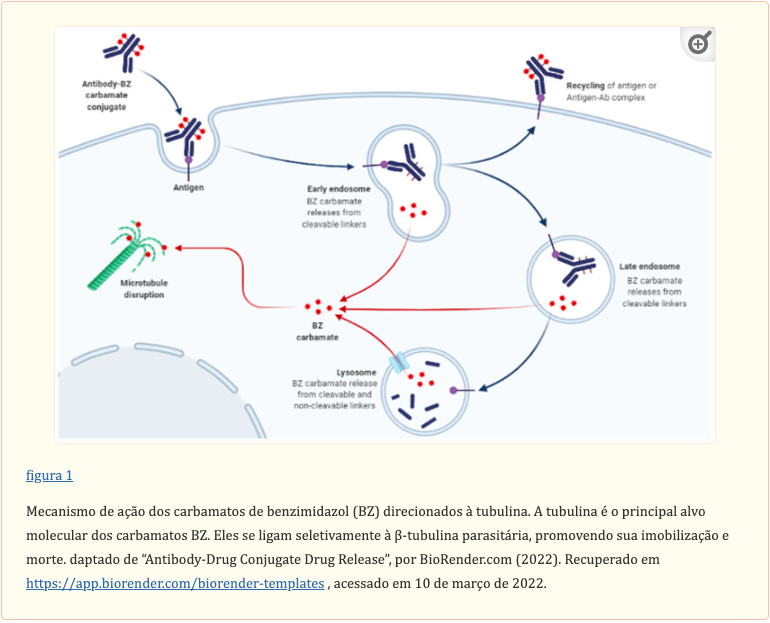
O modo de ação molecular dos carbamatos BZ envolve inibir a polimerização da tubulina e facilitar a ruptura dos microtúbulos nas células do parasita (figura 1). Um estudo in vitro usando os extratos de tubulina de helmintos e mamíferos implicou a tubulina como o principal alvo molecular dos carbamatos BZ. A tubulina é fundamental para a motilidade, proliferação e divisão celular; o transporte intercelular de organelas; a manutenção da forma celular; e o processo de secreção de células em todos os organismos vivos. Ao bloquear o alongamento dos microtúbulos em vermes, os carbamatos BZ perturbam a captação de glicose nas células. Eventualmente, as reservas de glicogênio são esgotadas e seus mecanismos de gerenciamento de energia são esgotados, culminando na morte dos parasitas.
3.2. Atividade anticancerígena de carbamatos BZ
Os carbamatos BZ são seletivos para células cancerígenas, causando citotoxicidade mínima em células normais, mas citotoxicidade aumentada em diferentes células tumorais. Vários estudos relataram que os carbamatos BZ inibem a polimerização da tubulina de mamíferos in vitro. Se o mesmo efeito seria observado em células humanas e, em caso afirmativo, se tais esforços direcionados poderiam ser eficazes contra tumores, são algumas questões levantadas por esses relatórios. Lacey et al. abordou pela primeira vez a atividade de carbamatos BZ contra células de leucemia de camundongo L1210 em 1985. Uma investigação mais completa sobre os efeitos antitumorais dos carbamatos BZ foi realizada; os resultados mais promissores desta investigação estão resumidos na tabela 1. As propriedades farmacocinéticas gerais dos carbamatos BZ são as seguintes: absorção lenta; ampla distribuição por todo o corpo; extenso metabolismo hepático; e excreção via urina e fezes (Figura 2a). Seus efeitos colaterais comuns são febre, náusea, vômito, desconforto abdominal e hepatotoxicidade. A baixa taxa de absorção intestinal dos carbamatos BZ pode tornar difícil para eles atingirem concentrações na circulação sistêmica eficazes no tratamento de cânceres em humanos. O aumento da biodisponibilidade é necessário para potencializar seu efeito antitumoral, tornando-os seguros e bem toleráveis no uso humano e veterinário.
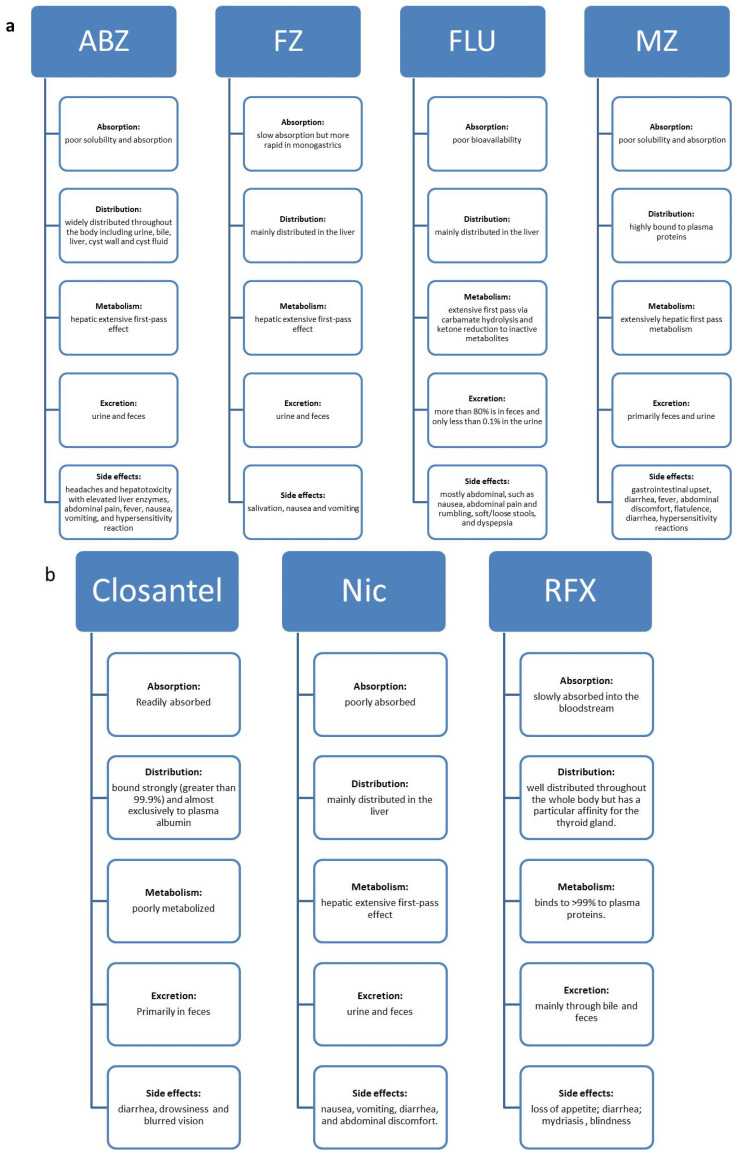
Figura 2
Propriedades farmacocinéticas e efeitos colaterais de medicamentos antiparasitários veterinários. (a) Os carbamatos BZ são pouco absorvidos; têm uma ampla distribuição no corpo; apresentam extenso metabolismo hepático; e são excretados pelas fezes e urina. (b) As drogas antiparasitárias salicilanilidas (HS) halogenadas apresentam má absorção, distribuição por todo o corpo, são pouco metabolizadas e são excretadas na bile, fezes ou urina.
3.2.1. ABZ
O ABZ é um medicamento aprovado pela FDA para o tratamento de vários tipos de infecções por vermes parasitários, como a doença hidática cística do fígado, pulmão e peritônio causada pela forma larval da tênia canina. Também tem sido objeto de investigações recentes como um agente anticancerígeno significativo, devido à sua toxicidade limitada para células normais, mas toxicidade de alto nível para células tumorais e parasitárias. Em 1989, a citotoxicidade do ABZ foi revelada na linha celular do carcinoma hepatocelular (HCC) Hep2G. Além disso, descobriu-se que o ABZ inibe a proliferação de linhas celulares HCC humanas, de ratos e camundongos. Estas observações in vitro foram então recapituladas in vivo pelos mesmos autores usando um modelo de xenoenxerto contra o desenvolvimento de células SKHEP-1 de tumor humano subcutâneo em camundongos nus, sugerindo o promissor potencial antitumoral do ABZ em humanos. A citotoxicidade ABZ foi examinada em diferentes linhagens celulares de câncer intestinal (SW480, SW620, Caco2, NCM460 e HCT-8) com diferentes origens e características de crescimento, representando vários estágios de malignidade. Pougholami et al. forneceu evidências de sua eficácia no tratamento da carcinomatose peritoneal decorrente de linhagens de células colorretais. Verificou-se que o ABZ e seu principal metabólito ABZ sulfóxido inibem acentuadamente o desenvolvimento e a proliferação da linha celular colorretal humana HT-29. O mecanismo por trás dos efeitos examinados dos compostos empregados foi a indução da parada do ciclo celular na fase G2/M e a ativação da apoptose mediada por caspase-3. Citotoxicidade significativa foi observada na linha celular de leucemia CEM/dEpoB300 resistente ao paclitaxel (PTX) tratada com ABZ. O ABZ estimulou com sucesso a despolimerização maciça da rede microtubular e a ativação da apoptose, mediada por uma profunda regulação negativa das proteínas BCL-2 e MCL-1. Também facilitou a despolimerização de microtúbulos em células de câncer de ovário 1A9PTX22 e foi considerado mais eficaz do que o PTX na inibição de sua proliferação. A baixa solubilidade do ABZ impede que ele seja absorvido em quantidades suficientemente grandes nos órgãos para ser tóxico para os seres humanos.
3.2.2. FZ
A atividade antitumoral de FZ também foi investigada em diferentes linhagens celulares. Os pesquisadores descobriram que FZ manifesta atividade moderada de despolimerização de microtúbulos em células cancerígenas humanas, como ficou evidente em experimentos in vitro e in vivo. FZ exibe seu efeito antitumoral através da ruptura de microtúbulos, ativação de p53 e modulação de genes associados a múltiplas vias celulares. O tratamento com FZ também culminou na redução da captação de glicose nas células cancerígenas, causando a regulação negativa dos transportadores GLUT e das enzimas glicolíticas essenciais. Qiwen et al. relataram que o tratamento com FZ foi tóxico para células mamárias EMT6 in vitro. O nível de toxicidade aumentou com o tempo de incubação e sob condições de hipóxia grave. FZ também aumentou os efeitos plásticos da radiação. Demonstrou uma afinidade moderada para tubulina de mamífero e exibiu citotoxicidade para linhas de células de câncer humano (H460 e A549) em concentrações micromolares. Além disso, causou a translocação mitocondrial de p53 e inibiu eficientemente a captação de glicose e a expressão de transportadores GLUT, bem como hexoquinase, uma enzima glicolítica chave regulada positivamente na maioria das células cancerígenas. Também bloqueou o crescimento de xenoenxertos humanos em um nu/numodelo de camundongos quando administrado por via oral. Notavelmente, FZ não mostrou nenhuma toxicidade óbvia para células epiteliais primárias de tecido pulmonar de rato. No entanto, Ping e cols. mostraram que o FZ sozinho não poderia afetar o crescimento das linhas celulares de linfoma humano P493-6 em camundongos SCID. No entanto, FZ em combinação com suplementação vitamínica preveniu significativamente o crescimento tumoral, indicando sua aplicabilidade em estudos antitumorais. Adicionalmente, o efeito de uma dieta terapêutica contendo 150 ppm de FZ, injetada por via intradérmica em camundongos BALB/c, no crescimento de tumores mamários de camundongos EMT6 foi examinado por Duan et al. Eles concluíram que FZ não alterou o crescimento do tumor, invasão ou metástase, e deve-se tomar cuidado ao administrar alimentos contendo FZ a colônias de camundongos para pesquisa de câncer.
3.2.3. GRIPE
A atividade anticancerígena da FLU foi investigada pela primeira vez em células de leucemia e mieloma originárias de linhagens de células estabilizadas estabelecidas e amostras de pacientes. As combinações de FLU com vinblastina ou vincristina retardaram o crescimento do tumor no modelo de xenoenxerto mais do que qualquer uma das drogas isoladamente. Os autores declararam o FLU como um novo inibidor de microtúbulos exibindo atividade pré-clínica em leucemia e mieloma. Em outras investigações, a FLU inibiu com sucesso a proliferação de várias linhagens de células-tronco de câncer de mama (MDA-MB-231, BT-549, SK-BR-3 e MCF-7) de maneira dependente da dose e do tempo, e atrasou o crescimento do tumor em modelos de xenoenxerto via injeção intraperitoneal. Além disso, a gripe mostrou um efeito anticancerígeno contra o câncer colorretal (CRC) e parecia parcialmente seguro para células normais.
3.2.4. MZ
Verificou-se também que o MZ é eficaz contra muitas linhagens de células cancerígenas. MZ inibiu significativamente a proliferação de células de carcinoma adrenocortical devido à indução de apoptose. Além disso, suprimiu a invasão e migração de células cancerígenas in vitro e a formação de metástases em animais experimentais. O tratamento de linhagens celulares de câncer de pulmão com MZ causou parada mitótica por inibição da polarização da tubulina seguida de apoptose. A administração oral de MZ em camundongos estimulou um efeito antitumoral robusto em um modelo subcutâneo e reduziu as colônias pulmonares em metástases pulmonares induzidas experimentalmente, sem mostrar qualquer toxicidade em camundongos tratados com PTX. É relatado que 80% das linhas celulares de câncer de cólon eram sensíveis a MBZ no painel NCI 60. MBZ causou remissão completa das metástases nos pulmões e linfonodos, sem nenhum efeito adverso aos pacientes. Além disso, MZ mostrou atividade anticancerígena contra câncer gástrico (GC), tumor cerebral, glioblastoma, meduloblastoma, câncer de próstata, câncer de ducto biliar, câncer pancreático, câncer de cabeça e pescoço, câncer de mama e melanoma. O MZ foi testado pré-clinicamente como um substituto para a vincristina, uma droga que tem toxicidade limitante da dose no tratamento de tumores cerebrais.
3.3. Atividade anticancerígena de carbamatos BZ em modelos clínicos
Um estudo piloto do efeito do ABZ em sete pacientes com carcinoma hepatocelular avançado e CCR com metástases hepáticas refratárias a outras formas de terapia foi realizado por 28 dias. Os pacientes receberam ABZ por via oral (10 mg/kg/dia) em duas doses divididas. Os níveis de marcadores tumorais, antígeno carcinoembrionário e a-fetoproteína foram medidos rotineiramente. Os parâmetros de índices hematológicos e bioquímicos também foram obtidos para monitorar as toxicidades medular, renal e hepática. Os resultados desta pesquisa confirmam ainda mais a tolerância ao ABZ em pacientes, com o único efeito colateral preocupante sendo a neutropenia aguda em três dos pacientes. Além disso, o ABZ reduziu significativamente dois marcadores tumorais em dois pacientes, enquanto em outros três pacientes os marcadores foram estabilizados, demonstrando que o ABZ possui efeitos antitumorais em humanos.
No subsequente ensaio de fase I de determinação da dose de ABZ oral em pacientes com tumores sólidos incontroláveis, 36 pacientes receberam uma dose inicial de 400 mg com aceleração da dose até 1200 mg duas vezes ao dia em ciclos de três semanas. Amostras de sangue sequenciais foram coletadas por até 96 horas e no dia oito dos ciclos um e quatro. Os autores concluíram que o ABZ foi bem tolerado para o esquema testado neste estudo e que a dose recomendada para estudos posteriores foi de 1.200 mg duas vezes ao dia por 14 dias em um ciclo de 21 dias.
Um paciente com câncer de pulmão de pequenas células experimentou FZ com suplementos de vitamina E, óleo de canabidiol e curcumina biodisponível durante um ensaio clínico. Uma tomografia por emissão de pósitrons após três meses não encontrou células cancerígenas no corpo do paciente. Esta história de sucesso, juntamente com outras 40 histórias de sucesso conhecidas da FZ, foi partilhada através de um serviço de rede social e um blog. Também vale a pena mencionar que o instituto de pesquisa Oklahoma Medical Research Foundation, em colaboração com Stanford e a Emory University, concordou em ajudar todos os pacientes com câncer usando FZ com uma revisão clínica do protocolo FZ.
Atualmente, vários ensaios clínicos de MZ em diferentes tipos de câncer estão sendo conduzidos. Um homem de 48 anos com carcinoma adrenocortical parou de tomar qualquer droga quimioterápica e foi prescrito MZ como agente único na dosagem de 100 mg duas vezes ao dia. A metástase do câncer no paciente inicialmente regrediu e, subsequentemente, permaneceu estável, sem apresentar efeitos adversos clinicamente significativos. Outro homem de 74 anos com câncer de cólon metastático foi prescrito MZ na dosagem de 100 mg duas vezes ao dia depois que outros agentes anticancerígenos, como capecitabina, oxaliplatina e bevacizumabe, resultaram em neuropatia intolerável induzida por oxaliplatina. O paciente não experimentou efeitos adversos do tratamento, com remissão quase completa das metástases nos pulmões e linfonodos e boa remissão parcial no fígado. Para investigar a segurança e a eficácia do MBZ com dosagem individualizada no câncer, 11 pacientes com câncer gastrointestinal avançado foram tratados com MBZ com dose ajustada individualizada de até 4 g/dia para atingir uma concentração sérica de 300 ng/mL. Cinco pacientes atingiram a concentração sérica alvo de MBZ sem efeitos adversos significativos. Verificou-se que o MBZ individualizado com dose ajustada é seguro e bem tolerado em pacientes com câncer em estágio avançado. Um estudo aberto de fase I em pacientes recém-diagnosticados com glioma de alto grau recebendo MZ em combinação com temozolomida não apresentou efeitos colaterais graves. Os pacientes receberam 500 mg de MZ três vezes ao dia em um ciclo de 28 dias. O objetivo principal era determinar a dose tolerada mais alta de MZ com temozolomida e investigar se essa combinação poderia retardar o crescimento de tumores cerebrais (NCT01729260). O segundo ensaio clínico nas fases I e II foi um estudo piloto de MZ em combinação com carboplatina, temozolomida e vincristina (NCT01837862). O MZ foi administrado a pacientes por 70 semanas na dosagem de 100 mg duas vezes ao dia. O objetivo principal da fase I foi verificar a tolerância do MZ em combinação com três agentes quimioterápicos atuais. A fase II monitorou a duração do estado livre de progressão em pacientes e sua sobrevida global. Além disso, para avaliar a segurança e eficácia do MZ, um ensaio clínico de fase II foi realizado em pacientes com câncer gastrointestinal ou câncer de origem desconhecida (NCT03628079). Todos os pacientes receberam tratamento contínuo com MZ por 16 semanas, e as doses individuais foram determinadas com base na concentração sérica de MZ. O estudo foi encerrado em janeiro de 2020, sem resultados relatados ainda. Além disso, o MZ, em combinação com metformina, doxiciclina e atorvastatina, tem sido usado no câncer para verificar a segurança, tolerabilidade e eficácia dos tratamentos combinados (NCT02201381). Além disso, um tratamento adjuvante de Fase 3 de MZ usado no câncer de cólon também foi realizado (NCT03925662).
No momento, nenhum estudo clínico sobre FLU e FZ em malignidades humanas foi realizado. Um relatório mais completo sobre ensaios clínicos documentando os efeitos antitumorais de drogas antiparasitárias é resumido tabela 2.
4. SS
As salicilanilidas são um grupo muito grande de compostos que apresentam atividade eficiente contra certos tipos de parasitas. Sua estrutura química básica consiste em um anel de ácido salicílico e um anel de anilida. Exemplos de drogas HS com potente atividade anti-helmíntica são Nic, rafoxanida (RFX) e closantel.
4.1. Mecanismo de Ação do HS
O principal mecanismo de ação do HS foi investigado in vitro usando mitocôndrias de fígado de rato e moscas domésticas. Os autores encontraram uma associação com o desacoplamento da fosforilação oxidativa que interrompe a produção de ATP. Isso parece acontecer através da supressão da atividade de duas enzimas, succinato desidrogenase e fumarato redutase, e assim prejudica a motilidade dos parasitas e eventualmente causa a morte. Vários pesquisadores posteriormente confirmaram o mecanismo proposto in vivo.
4.2. Atividade anticancerígena do HS
Várias drogas do grupo HS foram investigadas quanto ao seu efeito sobre o câncer em modelos experimentais e pré-clínicos. As propriedades farmacocinéticas e os efeitos colaterais comuns dos medicamentos HS são mostrados na Figura 2b.
4.2.1. Closantel
Closantel é outro agente salicilamida halogenado com potente atividade antiparasitária veterinária contra parasitas internos e externos, como lombrigas e vermes hepáticos. Foi relatado que inibe o BRAF V600E. Zhu et al. avaliaram a atividade antiangiogênica do closantel em peixe-zebra com IC 50 de 1,69 μM nos vasos intersegmentares e 1,45 μM nos vasos subintestinais. Closantel também proibiu notavelmente o crescimento do câncer em peixes-zebra xenotransplantados com linfoma humano, câncer cervical, células de câncer de fígado e câncer pancreático de maneira dose-dependente. Eles relataram que concentrações mais baixas de closantel não induziram nenhum efeito colateral notável. No entanto, investigações adicionais são necessárias para confirmar a atividade anticancerígena do closantel em humanos, a fim de fornecer novos insights sobre sua possível aplicação clínica.
4.2.2. Nic
Nic é um medicamento aprovado pela FDA amplamente utilizado para o tratamento de infecções por tênia. Vários estudos estabeleceram a atividade anticancerígena de Nic (5-cloro-N-(2-cloro-4-nitrofenil)-2-hidrobenzamida) em modelos in vitro e in vivo. Foi relatado que o Nic inibe a ativação e a função transcricional da sinalização de STAT3 e Wnt/β-catenina induzindo a degradação de LRP6 de forma eficaz e, como resultado, induz a supressão do crescimento celular, apoptose celular e interrupção do ciclo celular de células cancerígenas. Além disso, Nic não apresentou toxicidade significativa em células não cancerígenas in vitro e não revelou efeitos colaterais em camundongos tratados com Nic. Os efeitos inibitórios do Nic nas células-tronco cancerígenas (CSC) antecipam sua aplicação na terapia do câncer. Nic suprimiu o crescimento de células semelhantes a CSC e mecanicamente, com ou sem cisplatina, inibiu significativamente a transição epitelial-mesenquimal e o crescimento tumoral no câncer de mama, sem sintomas aparentes dos efeitos citotóxicos de Nic nos camundongos tratados. Foi relatado que Nic em combinação com TRA-8 suprime o crescimento de xenoenxertos tumorais em cânceres de mama basais, reduzindo a atividade Wnt/β-catenina.
Nic demonstrou interromper a interação de p65 com FOXM1/β-catenina e inibir a via NF-κB, erradicando assim as células-tronco da leucemia na leucemia mieloide crônica e na leucemia mieloide aguda, respectivamente. Além disso, o Nic inibiu várias vias de transdução de sinal, como a via Wnt/β-catenina, e também induziu a apoptose no câncer de cólon, isoladamente ou em combinação com erlotinibe, e pode funcionar como um agente terapêutico potente para pacientes com polipose adenomatose familiar (FAP). interrompendo o complexo Axin-GSK3. Desacopladores mitocondriais, como Nic etanolamina e oxiclozanida, demonstraram atividades anticancerígenas significativas para o tratamento de tumores metastáticos hepáticos em um modelo de camundongo, com base em sua atividade de desacoplamento mitocondrial. Recentemente, várias investigações descobriram que o Nic previne o crescimento de células de carcinoma ovariano, xenoenxertos ovarianos e múltiplas vias de sinalização metabólica, como as vias de sinalização Wnt/β-catenina, JAK2/STAT3 e mTOR, afetando a biogenética, a biogênese e a regulação redox em células iniciadoras de tumor de câncer de ovário. A citotoxicidade do Nic foi investigada contra HCC e células de câncer de pulmão humano, onde causou notável apoptose celular dependente da dose, suprimiu a viabilidade celular e inibiu a formação de clones. Além disso, Nic também mostrou citotoxicidade em células de glioblastoma humano, visando múltiplas vias de sinalização celular principais.
Nic demonstrou um potencial para direcionar o eixo de sinalização do receptor de andrógeno-STAT3, superando a resistência à enzalutamida, bem como suprimindo a migração celular e a invasão no câncer de próstata avançado. Chen et al. descobriram que o Nic inibiu notavelmente o alvo mamífero da rapamicina, a via de sinalização mTOR e a respiração mitocondrial em um painel de linhagens celulares de câncer cervical e sugeriu o Nic como uma potencial droga terapêutica para o câncer cervical. Nic direcionou células de câncer de esôfago resistentes a PTX in vitro e in vivo, por meio da inibição de Wnt/β-catenina, enquanto mostrava toxicidade mínima em relação a células normais. Também foi relatado que o Nic pode efetivamente impedir o crescimento do tumor em um modelo de tumor de xenoenxerto de camundongo de células de osteossarcoma humano, visando múltiplas vias de transdução de sinal. No carcinoma adrenocortical (ACC), Nic exibe potente atividade anti-ACC inibindo múltiplas vias celulares e danificando o metabolismo celular. Além disso, Nic proíbe a expressão de C-MYC e E2F1, enquanto induz a expressão de PTEN no carcinoma de células renais (RCC). Nic demonstrou ainda sinergizar com o agente de terapia direcionado Sorafenib na supressão do crescimento e sobrevivência de células RCC. Também demonstrou eficácia potencial ao inibir o enriquecimento de carcinoma oral humano de células escamosas induzido por cisplatina e aumentar a sensibilidade à cisplatina em esferas tumorais ALDH+.
4.2.3. RFX
RFX, N-[3-cloro-4-(4-cloro-fenoxi) fenil]-2-hidroxi-3, 5-diiodobenzamida, é outro composto salicilanilida halogenado que é usado para o tratamento de infecções causadas por Fasciola hepatica e alguns vermes redondos gastrointestinais. Embora o RFX seja aprovado pelo FDA para uso veterinário, poucas informações sobre seus efeitos terapêuticos em humanos estão disponíveis. Um estudo anterior relatou o uso terapêutico de RFX em uma menina de sete anos com fasciolíase. Mecanisticamente, o RFX foi relatado como um inibidor B-Raf V600E significativo que suprimiu a ativação da via p38 MAPK, induzindo a apoptose celular pela redução do potencial de membrana mitocondrial no mieloma múltiplo. O RFX não exibiu toxicidade em duas linhagens de células humanas em concentrações de até 12,5 μM em um estudo avaliando os efeitos do RFX na fase estacionária hipervirulenta de Clostridium difficile, e reduziu significativamente a atividade hemolítica em comparação com Nic.
Shi et al. investigaram a inibição da atividade de CDK4/6 por RFX e o sugeriram como um candidato promissor para o tratamento do câncer de pele humano. O estudo não observou nenhuma toxicidade perceptível ou alteração no peso corporal dos camundongos nus BALB/C administrados com REF. Além disso, a administração sistêmica de RFX a camundongos induziu apoptose e acelerou a inibição de células cancerígenas do cólon por meio da ativação da cascata ERS. Mais recentemente, estudos indicaram que o RFX é um indutor de morte celular imunogênica (ICD) bona fide em células CRC. Em termos gerais, induziu todos os principais padrões moleculares associados ao dano (exposição à ectocalreticulina e liberação de ATP/grupo de alta mobilidade caixa 1) essenciais para o CDI. In vivo, o RFX reduziu notavelmente a proliferação tumoral em células CRC HCT-116 e DLD1 CRC em comparação com o falso. Os efeitos anticancerígenos do RFX também foram revelados em GC, onde suprimiu a proliferação de células GC na fase G0/G1 do ciclo celular e estimulou a apoptose e a autofagia através da inibição da via PI3K/Akt tanto in vitro quanto in vivo.
4.3. Atividade anticâncer de HS em modelos clínicos
Nic passou por ensaios clínicos em pacientes com câncer de cólon ressecável em 2017, mas foi encerrado devido à baixa taxa de inscrição (NCT02687009). Dois outros estudos clínicos estão atualmente em andamento para testar os efeitos anticâncer de Nic em pacientes com PAF (NCT04296851) e progressão de metástases de câncer colorretal após a terapia (NCT02519582). Embora um estudo de fase I de Nic administrado junto com enzalutamida em pacientes com câncer de próstata resistente à castração tenha sido concluído, antecipando o início de um estudo de fase 2 (NCT02532114), outro ensaio clínico de fase I está investigando a dose potente e os efeitos colaterais de Nic em combinação com enzalutamida para tratar pacientes com câncer de próstata resistente à castração (NCT03123978). Um ensaio clínico de fase II também está em andamento para avaliar a eficácia do acetato de abiraterona, Nic e prednisona no tratamento de pacientes com câncer de próstata resistente a hormônios (NCT02807805).
5. Discussão
O reaproveitamento de medicamentos aprovados e abandonados tem ganhado enorme atenção no desenvolvimento de medicamentos oncológicos, representando uma solução para o custo exorbitante de descoberta de novos medicamentos. Além disso, a aprovação pode ser acelerada pelo conhecimento de dados pré-clínicos e clínicos sobre farmacocinética, toxicidades e regimes. Existe um interesse nas atividades anticancerígenas de vários agentes antiparasitários devido aos seus efeitos colaterais menos graves em comparação com os quimioterápicos convencionais (principalmente agentes que danificam o DNA) na qualidade de vida dos pacientes. Carbamatos BZ e drogas HS, quando administrados sozinhos ou em combinação, provocam atividades antitumorais demonstradas por várias ações biológicas, como indução de apoptose e autofagia; redução da viabilidade celular, migração e invasão; interromper a polimerização da tubulina; induzindo diferenciação e senescência; redução da angiogênese; prejudicando a utilização de glicose; interrompendo o ciclo celular; e visando várias vias de transdução de sinal oncogênico chave. Esses agentes induzem citotoxicidade mínima em células normais, mas citotoxicidade comparativamente alta em células tumorais, exercendo seletividade específica para células cancerígenas.
No entanto, os ensaios clínicos continuam sendo o principal gargalo para o reposicionamento bem-sucedido de agentes antiparasitários veterinários em oncologia, porque dados suficientes e úteis nem sempre estão disponíveis. Outro impedimento para o reposicionamento bem-sucedido de medicamentos antiparasitários veterinários como agentes anticancerígenos está relacionado às suas propriedades físico-químicas, via de administração e baixa biodisponibilidade. Embora a segurança dos carbamatos BZ e medicamentos HS para cuidados veterinários tenha sido avaliada, seu uso como agentes anticancerígenos não foi bem estabelecido para aplicação humana. As atividades potenciais, como toxicidade, efeitos colaterais e eficácia, dos medicamentos reaproveitados em populações de células cancerígenas e não cancerígenas após administração de curto e longo prazo garantem investigações experimentais aprofundadas no futuro.
Apesar dos dados limitados, alguns desses medicamentos já foram submetidos a ensaios clínicos de fase II/III para a terapia do câncer. Alguns pacientes com câncer metastático em estágio avançado responderam bem a ABZ, MZ e Nic, mostrando marcadores tumorais e metástases diminuídos e progressão estabilizada da doença. Ensaios clínicos em andamento fornecerão mais clareza sobre o potencial dos carbamatos BZ e drogas HS para uso em tratamentos de câncer primário e metastático em um futuro próximo. Há também evidências de que muitos pacientes desesperados estão tomando medicamentos antiparasitários sem informar seus médicos. Quando um paciente prescreve medicamentos antiparasitários em combinação com um medicamento anticancerígeno estabelecido durante um ensaio clínico, o resultado do ensaio clínico pode ser alterado; isso pode resultar em enormes perdas econômicas e no desperdício de um tempo precioso. Nesses cenários, as razões para os efeitos terapêuticos ou os efeitos adversos do medicamento em estudo no ensaio clínico permanecerão ambíguas. O especialista em câncer ficará confuso por não saber da administração oculta do medicamento antiparasitário auto-prescrito. Para evitar isso, os ensaios clínicos de drogas antiparasitárias precisam ser priorizados. Informações sobre a evidência de atividade antitumoral e a ausência de toxicidade na administração a longo prazo dessas drogas podem facilitar o caminho para futuros ensaios clínicos e determinar o melhor regime posológico.
Portanto, argumentamos a favor desses compostos, dada sua farmacocinética bem estabelecida, excelente perfil de toxicidade e baixo custo. Acreditamos que uma entrevista primária de pacientes com câncer, juntamente com pesquisadores interdisciplinares, como veterinários, especialistas em câncer e farmacologistas, valerá a pena para facilitar os ensaios clínicos de medicamentos antiparasitários veterinários com uso potencial para pacientes com câncer.
Fonte: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9029030/